EU MDR vs FDA: what are the main differences and similarities?

What is the best approach to take when expanding to the EU market from the US and vice versa? To what extent do the MDR and FDA overlap and what are the key differences and similarities between them? How can you plan an effective strategy for entering new markets that allows you to avoid extra costs and reduces effort?
The different regulatory requirements between the EU and US markets can be a significant stumbling block for medical device manufacturers who plan an expansion across the pond. In this article, prepared in cooperation with Krzysztof Minicki, our Director of Healthcare and Life Sciences, we compare the EU MDR and FDA and discuss the optimal approach for releasing a medical device to both markets.
What are the differences and similarities between the EU MDR and FDA regulations?
There are several differences between the FDA and MDR requirements, but also some similarities in certain areas. Important ones are listed below.
Our experts will help you prepare a Technical File
Find out more
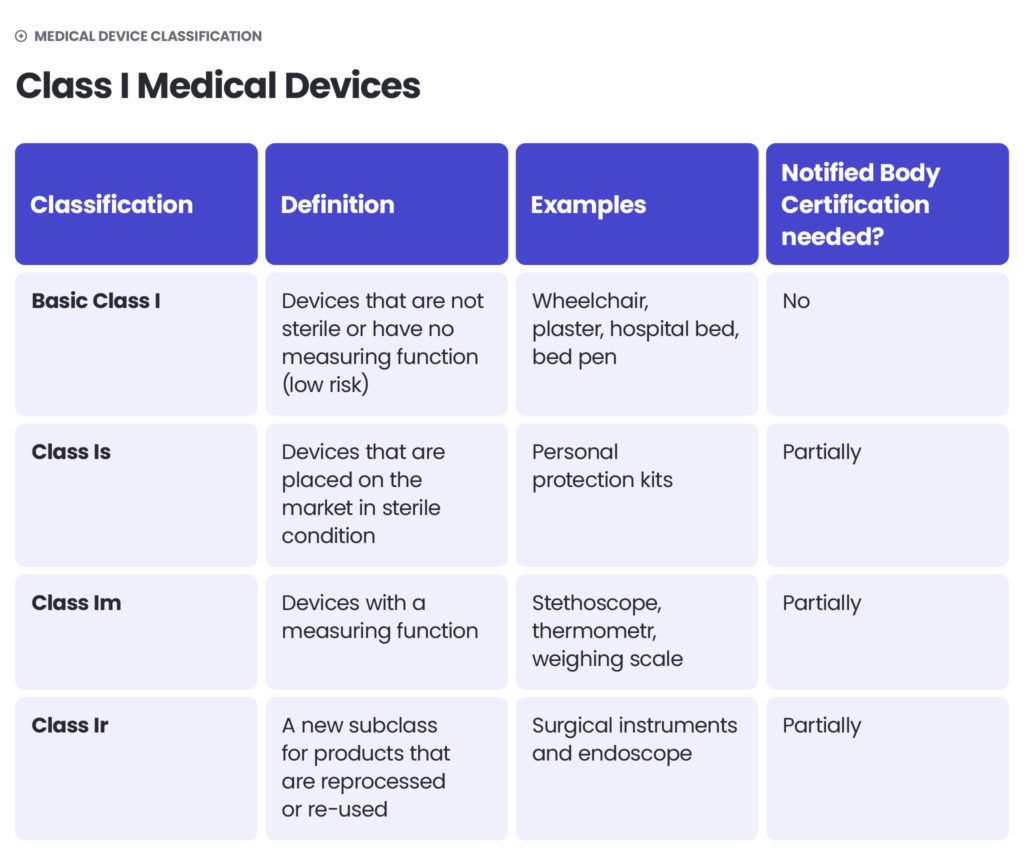
Medical device classification
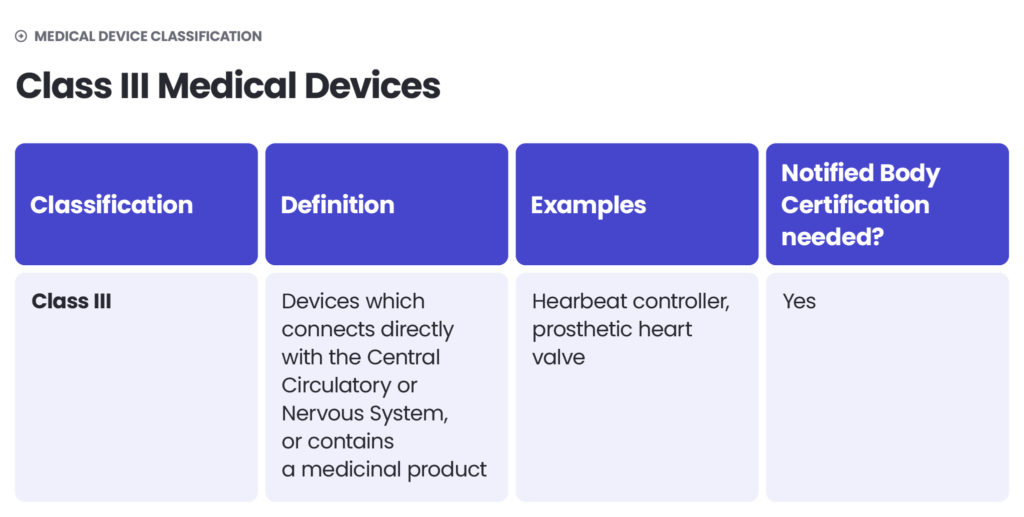
Under the EU MDR, there are classes I, II (further divided into IIa and IIb) and III. The higher the class – the higher the risk. Class I includes medical devices of low risk to users (non-sterile and non-measuring devices) as well as devices of a slightly higher risk, but which are non-invasive (sterile and measuring devices). Class IIa includes middle-risk devices and class IIb – middle- to high-risk devices. Class III devices pose the highest risk to patients or operators. They are described as those which connect directly with the Central Circulatory or Nervous System or contain a medicinal product.
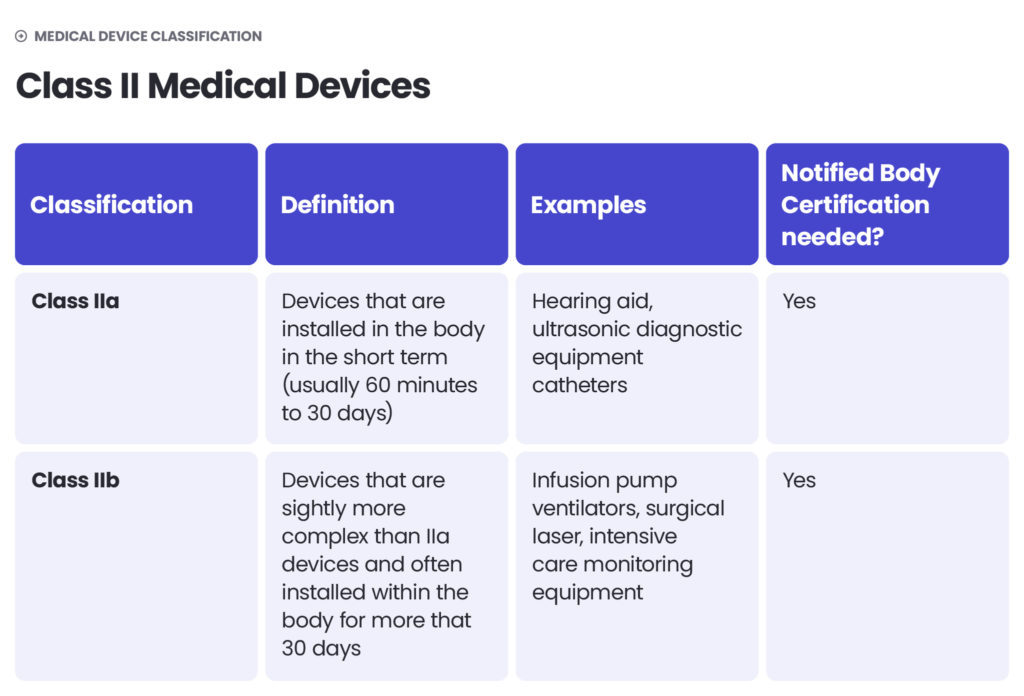
The approach to medical device classification under the FDA is quite similar to the MDR. The FDA also classifies medical devices according to their risk level and regulatory controls necessary to ensure patient and operator safety: class I is associated with the least risk, class II with moderate risk and class III with the highest risk.
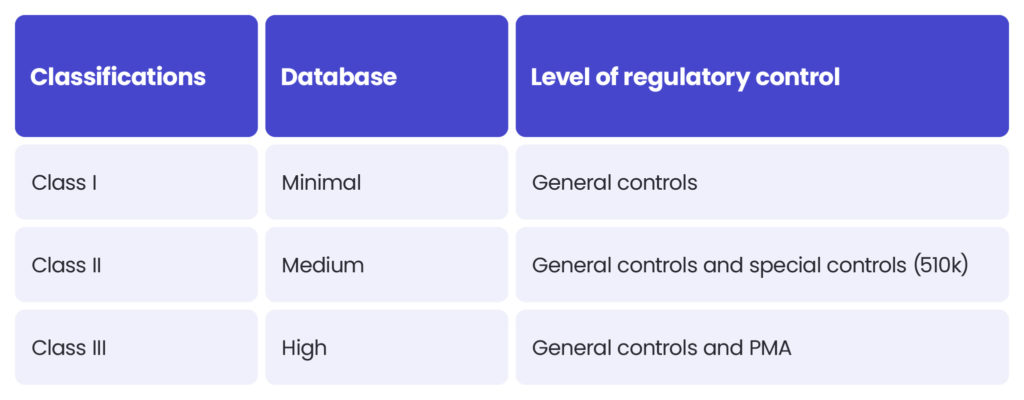
However, there may be differences between the MDR and FDA in assessing the risk level. The MDR Class I sterile or measuring devices may be regarded as class II under the FDA’s regulation. Certain MDR class IIb medical devices could be regarded as class III according to the FDA. Each case should be assessed independently.
Recommended reading:
Quality management systems
Both the MDR and FDA require manufacturers of medical devices, including software, to have a Quality Management System implemented that is compliant with respective regulations.
The scope of content
The FDA differs from the EU MDR in scope and how the information is organised. The FDA regulations are divided into sections according to the categories of medical devices. Each section includes requirements regarding the safety and effectiveness of medical devices as per their use. It’s directly connected to the premarket submission method followed later in the process.
The MDR focuses more on the specific requirements for medical device manufacturers and notified bodies who overview the process of introducing new medical devices to the EU market. The MDR also includes many definitions and detailed information on financial liabilities and penalties, which aren’t within the scope of the FDA regulations.
Submission process
According to the FDA and MDR, class I medical devices (of the lowest risk, non-measuring and non-sterile ones) are not required to undergo an independent audit before their release to sale. To introduce them to the EU and US markets, the manufacturers must follow a self-declaration process (in the EU, it’s a CE-marking process). Naturally, all the appropriate documentation must be prepared, but third-party verification is not required.
Medical devices of higher risk must be verified either by a notified body in the EU or by the FDA in the US. A Technical File must be prepared in both cases, even though the release-to-market process is different.
Recommended reading: How to get FDA approval for medical devices?
The most common submission method under the FDA regulations is a Premarket Notification 510(k). Once you deliver all the necessary documentation, the FDA audits and verifies your submission. Under the EU MDR, the audit is related to the review of the Quality Management System, which is regulated by ISO 13485. The norm is harmonised, which means that it specifies certain legal requirements that need to be applied to achieve compliance . In other words, the compliance requirements can only be met if the norm is implemented in your organisation.
A medical device manufacturer who wants to introduce their product to the EU market for the first time needs to get certified in ISO 13485 beforehand. The certification process includes a two-stage audit. The first stage is the review of the processes and procedures related to the Quality Management System’s compliance with ISO 13485 and associated regulations (such as IEC 62304, ISO 14971, etc.). In the second stage, the technical documentation, including the Technical File of a medical device is reviewed to ensure its compliance with appropriate processes. However, under the MDR the review is only selective, while FDA conducts a full audit.
As per both the MDR and the FDA regulations, the medical device manufacturers are required to maintain control and ensure that a medical device will continue to be safe to use according to its Intended Use after it’s been released to market.
Medical Device Databases
There are different medical device databases functioning in the EU and the US, where all the information about the class of a medical device, its Intended Use, etc. are stored. In the US, it’s the FDA’s medical device database and in the EU – EUDAMED. They are separate and independent from each other.
Unique Medical Device Identification System
UDI is a globally accepted system and applies in the EU as well as in the US. The MDR expanded the concept of UDI by introducing the UDI-DI identification that allows for the grouping medical of devices that have the same intended purpose, risk class, essential design and manufacturing characteristics. However, there are certain differences between the MDR and FDA as to where and how the labels should be placed on medical devices.
Technical documentation
Despite the fact that the FDA and MDR regulations regarding the Technical File bear certain similarities, it doesn’t mean that the Technical File created for the EU requirements will be equally acceptable in the US and vice versa. Some differences lie in the nuances regarding manufacturing medical devices, such as risk management and classification.
For example, the FDA requires additional classification that defines Level of Concern. It doesn’t exist in the MDR regulation. It may be an obstacle for companies who already sell their medical devices on the EU market and now want to introduce their products to the US market.
What’s the right approach in such a case? When preparing technical documentation, you should include all the markets where you plan to sell your medical device. The market-specific information, for instance, regarding the aforementioned Level of Concern, should be included in the appropriate chapters of the documentation.
In other words, your documentation should cover the requirements specified in both the FDA and MDR. It’s not only the best possible, but also recommended, approach.
Clinical evaluation
For all class III and some class IIb medical devices, the EU MDR requires a Clinical Evaluation Report (CER) to be prepared by a manufacturer and then reviewed and accepted by a Notified Body. In contrast, the FDA doesn’t require CER for most medical devices qualified for a 510(k) submission.
The usability of medical devices
Both the FDA and the EU MDR put a strong focus on the usability of medical devices. Under the FDA, this concept is reflected by the Human Factors Studies. Human Factors Studies consist of two stages, formative and summative, and its goal is to check the usability and safety of software as a medical device (SaMD). The formative stage takes place at the beginning of the software development process, when wireframes and a proof of concept are created. The summative stage takes place at the end of the development process.
Our experts will help you prepare a Technical File compliant with the FDA and MDR
Preparing a single Technical File, compliant with both the FDA and MDR upfront, will save you extra costs compared to preparing a separate Technical File per each regulation. It’s worth planning the target markets for your medical device ahead of time.
Our consultants will assist you in preparing all the necessary technical documentation to meet the requirements of the EU and US medical device market. We’ll also help you get certified in ISO 13485, ISO 14971 or IEC 62304.
See our Healthcare and Life Sciences offering page for more details or contact Krzysztof Minicki directly using the form below.
The key difference lies in their regulatory frameworks – the EU Medical Device Regulation (MDR) is governed by European legislation applicable across all EU member states, while the U.S. Food and Drug Administration (FDA) operates under U.S. federal law. MDR emphasises documentation and post-market surveillance, whereas the FDA focuses on premarket approval processes and clinical performance.
The EU MDR categorises medical devices into four risk-based classes: Class I, IIa, IIb and III, with increasing regulatory scrutiny. The FDA uses three classes: Class I (low risk), Class II (moderate risk) and Class III (high risk). Although both systems are risk-based, the classification criteria and resulting regulatory pathways differ considerably.
Both regulatory systems prioritise patient safety, product performance and risk management. They require manufacturers to conduct clinical evaluations, implement quality management systems and carry out post-market surveillance. Additionally, both systems support a lifecycle approach to device regulation.
In the EU, conformity assessments are conducted by Notified Bodies, which are independent organisations designated by member states. In the US, the FDA directly reviews and approves or clears medical devices, particularly through pathways such as 510(k), PMA or De Novo classifications.
Under the EU Medical Device Regulation (MDR), clinical evaluation is an ongoing process throughout the device lifecycle, with a particular focus on real-world clinical data and post-market clinical follow-up (PMCF). By contrast, the FDA places greater emphasis on pre-market clinical trials, particularly for high-risk devices under the PMA pathway.
About the author
Malgorzata Kruszynska
Business Researcher
Contact us