How to get FDA approval for medical devices

The Food and Drug Administration (FDA) regulates the sale of products determined as medical devices and monitors their safety and effectiveness in the U.S. Therefore, to legally sell medical devices in that market, it is important to seek the agency’s approval. FDA approval means that the medical device was formally validated and that it is endorsed by the regulatory agency.
To answer the question about how to get the FDA approval for medical devices, we need to take a closer look at the whole process. Thus, in today’s article we introduce the most important parts of the process, such as device certification and selection of a suitable pathway, and we also give an overview of the costs and duration of the process. The publication was prepared in close collaboration with Krzysztof Minicki, Spyrosoft’s Director of Healthcare & Life Sciences.
FDA Approval process for Medical Devices
To get approval for your product, you need to go through a certification process and one of the approval pathways, while complying with a set of key regulations and requirements. The whole path to FDA Approval for Medical Devices can be described as a process of five steps:
Step 1 – Certification of the device
The first step to getting FDA approval is to properly classify your medical device. This step is especially important because the development process differs depending on the device’s classification. Medical devices are classified based on the risk they pose. FDA uses three types of classification:
- Class 1 – Devices pose the least amount of risk to consumers. Low-risk devices are subject to general control that ensures the safety and effectiveness of devices once they’re manufactured.
- Class 2 – Devices pose more risk to consumers than the ones in Class 1. Those devices are subject to both general and special controls. Special controls in this classification include labelling requirements, specific mandatory performance standards and specific testing requirements.
- Class 3 – Devices are made to support or sustain life, are implanted in the body, or have a high risk of illness or injury. Therefore, they require premarket approval, which means that producers must prove the safety and efficiency of the device. Besides premarket approval, Class 3 is also subject to general controls.
The classification of the device should be known before the development process begins. The class of a device can correlate with the way requirements and tests are managed. It also determines which regulations the device must comply with.
Need support with the FDA approval process?
Check how we can help
Step 2 – Creating a prototype
Next step in the approval process is to develop a prototype or an early version of the medical device. Such a device is not for human use, it is intended for tests made by researchers in a controlled laboratory environment. This stage provides important information about a product’s potential use and helps to identify and address its latent risks.
Step 3 – Pathway to Approval
Step three is the application process for the device, which is also dependent on the previous certification. FDA established the risk-based classification for medical devices, with three already mentioned regulatory classes. In which, as the device class increases, so does its regulatory control.
Since there are a few variants of device classification, manufacturers can choose from several application methods. The basic pathways to medical device approval are:
Premarket Notification 510(k)
This pathway is the most common one when launching a medical device. It is required for almost all Class II devices and some Class I devices. The purpose of this submission is to deliver documentation and evidence that a medical device is substantially equivalent in terms of safety and effectiveness to a device or devices that already legally exist on the market. Documentation must prove that both devices share the same intended use and technological characteristics, and also compare both in terms of design control process and verification testing.
Premarket Approval (PMA)
The PMA pathway covers Class III devices and those devices from Class I and Class II that couldn’t provide substantial equivalence in the 510(k) process. This process is the most complicated because it requires scientific evidence and clinical trials to prove the safety and effectiveness of the medical device.
Humanitarian Device Exemption (HDE)
HDE pathway is used in case of devices made to diagnose and treat diseases or conditions that affect a small amount of the population. A small number of patients results in difficulties in gathering enough clinical evidence to meet typical FDA standards. Therefore, in this process FDA has to grant Humanitarian Use Device (HUD) exemption, and then producers of the device must submit an HDE application to the adequate review centre. This pathway was prepared by the FDA to make sure that the devices seen as crucial for patients with rare conditions are subject to proper review and can be put on the market.
It is worth mentioning that in the situation where it’s difficult to find comparable devices already existing on the market, producers can use the De novo pathway option. This pathway requires a special petition to reclassify low- or moderate-risk devices as de novo ones. With that classification, the device can undergo a 510(k) application rather than a stricter PMA process. Devices approved in the de novo process can then serve as predicates for other devices.
Step 4 – Device review and approval
The next step is to wait for the FDA to review and approve the device. The review process starts when all information needed about the device is collected and complete. Some cases are exempt from the survey, but most of them are usually the subject of appropriate review, which depends on the classification previously assigned and types of similar devices already known on the market.
Premarket Notification 510(k)
Review of a Premarket Notification application usually doesn’t require clinical trials. It indicates whether the device is comparable to others that are already regulated and commercialised on the market. The 510(k) application must contain documented evidence that the product is similar to other devices and that it is substantially equivalent in safety and efficacy to the predicate device. Only after receiving a declaration that the device is substantially equivalent, can the producer launch the product in the U.S.
To determine if the device is substantially equivalent, FDA compares it to the predicate device in terms of:
- Having the same intended use and technological characteristics.
- Having the same intended use and different technological characteristics, while not, raising different safety and efficacy concerns. Moreover, the information submitted to the FDA shows that the device is as safe and effective as a legally marketed one.
Premarket Approval (PMA)
The submitted PMA application must include data from both clinical and non-clinical studies, which are the basis for the FDA experts’ decision on whether a device is safe and effective for market use. During the review and approval process, FDA focuses on things like:
- Administrative and scientific review to determine whether the proposal is complete.
- Conducting an in-depth scientific, regulatory, and quality review.
- Doing a review and possibly issuing further recommendations by the advisory committee.
- Final deliberations and notification on review outcome and final decision.
Humanitarian Device Exemption (HDE)
To submit the application for HDE, the producer must first receive an HUD exemption. With an HUD exemption, the FDA prepares an acceptance review to determine whether the application is complete and then conducts a substantive review. In this process, the agency checks if there is no other way to bring such a HUD to market and verifies whether there aren’t any other comparable devices on the market. To ensure this, the FDA considers the following:
- What are the device’s indications for use and technological characteristics?
- What is the population of patients that will be treated or diagnosed by the device?
- Whether the device fulfils the needs of the patients.
When necessary, the FDA may consult advisory committees, which are composed of independent experts on a given subject. The committee makes a recommendation on whether a device should be approved. With such a recommendation, the FDA decides and gives feedback to the submitter or requests for more information. Importantly, the final decision of the FDA must be published with all evidence in the Federal Register.
Step 5 – FDA Post-Market Device Performance Monitoring
After giving approval, the FDA continues to supervise the performance of the device to make sure that it’s effective and in line with all new safety concerns. The FDA conducts both announced and unannounced inspections in production facilities across the U.S. These audits can be routine or prompted by specific issues and are intended to ensure that producers are following good practices. If standards are not met, the FDA has the authority to close the facility.
To help report problems with already approved medical devices, FDA provides two programs:
- MedWatch – is used to report adverse events related to medical products (medicines and devices) and to learn about new safety information. FDA provides regular MedWatch safety alerts.
- Medical Product Safety Network (MedSun) – is used to report adverse events and to monitor the safety and effectiveness of medical devices by a group of 350 healthcare providers. The FDA publishes the MedSun newsletter once a month with key updates on medical device safety.
How much does it cost to get FDA approval for a medical device?
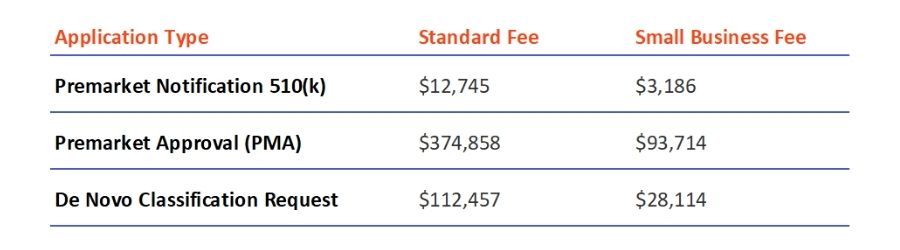
For most of its applications for medical device review, the FDA charges a fee that must be paid to start the approval process, unless the applicant is eligible for exemption. Besides the application costs, the FDA charges all businesses interested in launching their devices in the U.S. with an Annual Establishment Registration Fee, which is $5,672 for 2022.
The costs of the new fees are announced for each fiscal year in the Federal Register, and information about them can be found on the MDUFA user fee page. It is worth mentioning that for small businesses there is a chance of qualifying for reduced fees. The fees for the types of applications listed in the text are as follows:
How long does FDA approval take for medical devices?
The time it takes to get a medical device approved depends on its class and can last from one or two weeks to eight months.
Class I devices
Devices identified as Class I usually have the fastest path to the approval process. The majority of Class I devices can be self-registered and listed with the FDA. The process involves three phases – paying the registration fee, submitting listing and registration info, and receiving the acceptance email, all of which takes about a week or two.
A small percentage of Class I devices require 510(k) submission, the process that is normally used in the case of Class II devices.
Class II devices
The majority of Class II devices and some of the Class I devices require 510(k) application, which makes the approval process a bit longer. If the application is complete and there are no review issues, then the entire process can take around 90 days. However, if there are problems in the process or gaps in the documentation, the approval process can be significantly longer.
Class III devices
Products defined as Class III devices mostly follow the PMA pathway, which is known as the most complicated and stringent type of application. First, the FDA has 45 days to determine whether the application is sufficiently complete and to notify the applicant about the result of that verification. Then the FDA has 180 days to perform the PMA review and to approve or reject the application. Again, if the Agency identifies inconsistencies or shortcomings in the application that need to be filled, or if the applicant submits new relevant data or studies, the time of approval process may even double.
The class of the device and approval pathway have a significant impact on the length of the whole process. To shorten it, it’s important to write a good quality submission and to demonstrate substantial equivalence well or prove safety and efficacy of the device.
In need of support with the FDA approval process?
If you have further questions or need any assistance with the approval process, feel free to contact our Healthcare and Life Sciences team. Our experts can walk you through the whole process and help prepare needed documentation or choose the right certification class and submission pathway.




arrow_circle_rightCONTACT US