EU MDR: everything you need to know about Medical Device Regulation

May 2021 brought monumental changes to the way medical devices are classified. Up until then, companies were using regulations based on the Medical Device Directive implemented in 2017, which has been an indispensable document for medical classification. In May 2021, the Medical Device Regulation (EU MDR) were put in place, with their initial implementation due on May, 26th 2020 and then pushed back by a year due to the Covid-19 pandemic.
In today’s article, we’ll discuss the basics of EU MDR and we’ll look at how these changes influence the software development process.
What does the new EU MDR mean for the medical sector?
Before we start, it’s important to mention that these changes apply to the European Union market exclusively. The US, Canada, Japan and China have their own regulations that – to a limited extent, but differ from the European guidelines.
EU MDR provides stable, clear and balanced rules and a regulatory framework that is recognised internationally. It was introduced to increase clinical safety and unify the access to the medical market for manufacturers.
Before we start analysing the details of the EU MDR regulations, let’s go through the definitions that will be necessary for understanding them.
EU MDR – definitions
What is a medical device?
A medical device is an element, tool, application, software or an implant, manufactured to be used as a standalone item or together with other devices, and that is to be used for one or more activities on the following list:
- treatment, diagnostics, prevention, monitoring and prediction of diseases,
- any activity that’s aimed at supporting people with disabilities,
- examination, replacement and modification of anatomy, physiology and physiological processes in the human body, or any elements that affect metabolism, both directly and indirectly.
It is worth noting that devices for cleaning, disinfection and sterilisation fall under the latter category.
What is a medical accessory?
One of the elements that could affect a medical device and is regulated in a separate chapter of the MDR, is a medical accessory. The difference between a medical device and a medical accessory is that the latter will not be used for any activity from the list above, but it’s an important part of a medical device. It allows a device to work, supporting it within its Intended Use.
As Krzysztof Minicki, Healthcare and Life Sciences Business Unit Director, claims:
Speaking from my experience, software is often perceived as a medical accessory, while – according to the MDR – it’s an active medical device.
If we have a medical device with multiple functions to be turned off/on while configuring it or we have an application for configuring this device, then we can treat software as a medical accessory, but only if the former device can serve as a standalone instrument. An accessory is designed to support the activity and functionalities of a medical device.
What is an active medical device?
Active medical devices are defined as any device that provides energy to the human body and/or retrieves it. One of the examples is a radiograph.
It’s important to remember that MDR software is treated as an active medical device. It has a lot of implications for software manufacturers as it has extended the requirements for medical software.
What is a system according to the MDR?
Within the MDR, a ‘system’ indicates a set of devices that may or may not be packaged as a group, but it has one specific medical use and purpose. It’s a combination of multiple elements or devices that will work as a system.
What is a classification compliance assessment?
The medical classification was introduced to be used for marking medical devices with the CE Mark. This Mark means that a device has been properly classified and any risks associated with it have been assessed. Depending on the medical class, this compliance will be assured by a notified body – especially for classes IIa, IIb and III and for some devices in class I.
It’s worth mentioning that for your product to be CE marked, you need to go through the classification and compliance process. If your device is in one of the higher classes you have to go through the process with a notified body, and if your device is in class I, you can complete it on your own.
For some devices from classes II and III, there’s a new procedure based on a clinical assessment consultation, which is conducted by an independent expert panel based on the summary of a clinical assessment from a notified body related to the safety, functionality and technical documentation for the assessed device.
What is an Intended Use?
Intended Use is a document that defines the predetermined usage of a certain medical device that is then used to prepare a certification strategy and assess the class of said product.
One of the examples could be a device where one producer supplies it to laboratories with no intention of it being used as a medical device. Another producer could be manufacturing the same device, but with the Intended Use of surgeries performed at hospitals. The same device will then be classified as a medical device or as a non-medical device, depending on the Intended Use.
Let’s now discuss classes for medical devices used in the European Union.
What medical device classes are used in the EU?
There are four medical device classes currently used in the European Union: I, IIa, IIb and III.
It’s important to separate the classification of a device from the classification of software that may be a part of this device. Software, regardless of whether it’s a part of a device or a standalone product, needs to be classified separately. Additionally, the software also always needs to be classified using the IEC 62304 standard, where classes are marked as A, B and C.
How are medical devices classified?
MDR has 22 rules, applicable at different levels depending on the class of the risk. These rules determine how the classification should be conducted.
Technical documentation in the MDR
Before we go through any medical device assessment, we need to ensure that we have a number of documents and information about the medical device to be classified. The basic ones that need to be prepared and collected before the classification process, are:
- device description with its Intended Use, preferred usage and an end user group,
- data on the latter described as patients, and on the diseases that can be affected by the device, their treatment, diagnostics and monitoring.
What is a medical device classification?
Now, let’s move to the medical device classes.
EU MDR Class I Medical Devices
Class I is associated with the least risk, and any devices from this class cannot be invasive and they cannot be in direct contact with a patient. We treat this class as basic, in the Medical Device Directive (the predecessor of the MDR), software was always in class I.
Interestingly, class I devices don’t need to go through classification processes conducted by a notified body. Manufacturers can conduct them on their own. After completing the documentation and submitting it to the MDR system, the application will automatically be accepted after 14 working days.
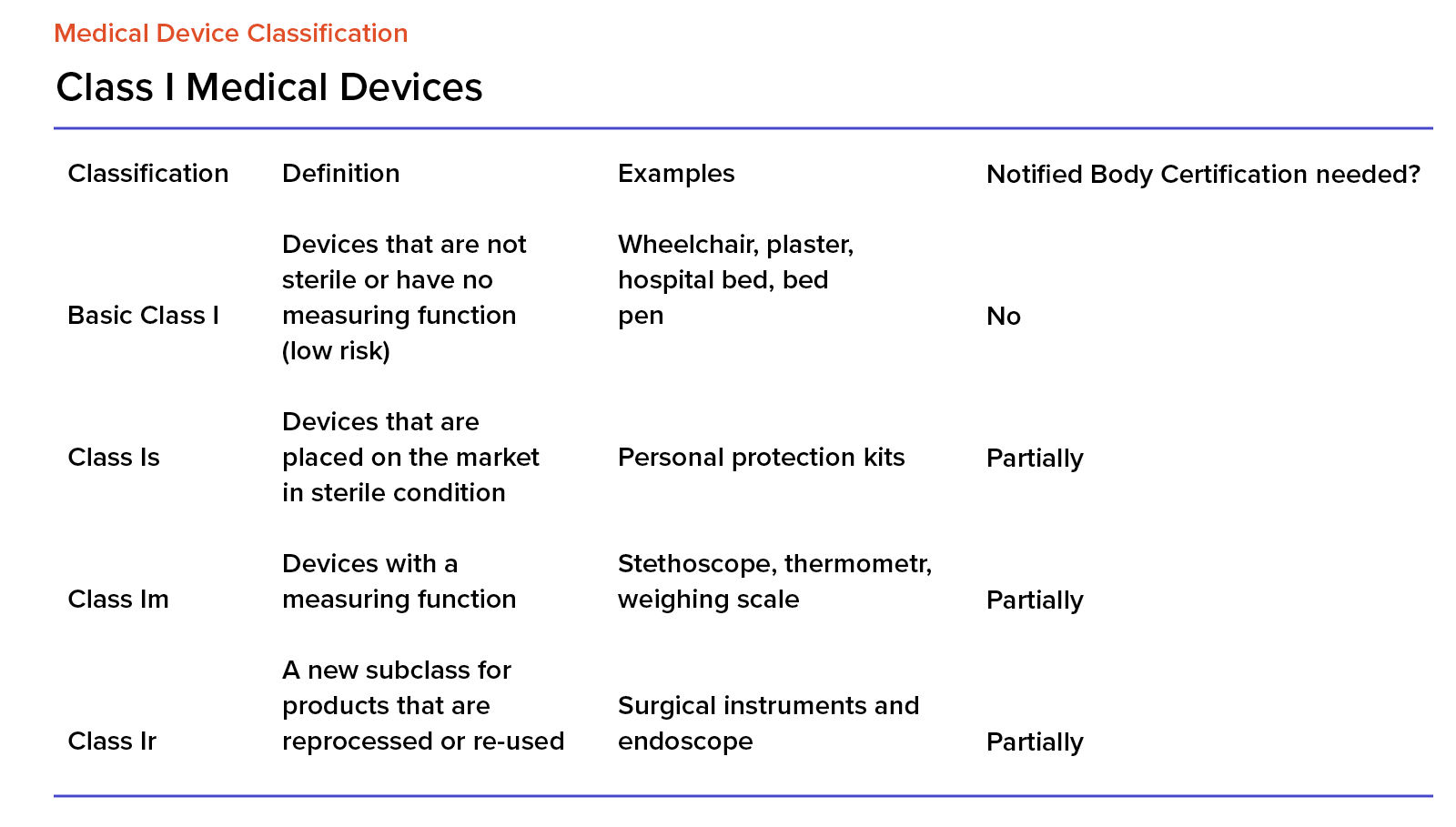
For medical devices in class I, we also have the following marks and designations:
- for sterile devices, it’s ‘s’ (as in ‘sterile),
- for measuring devices, it’s ‘m’ (as in ‘measure’),
- For devices that can be reused, it’s ‘r’ (as in ‘reuse’).
According to the data released by BVMED in 2017, 70% of medical devices are in class I.
EU MDR Class II Medical Devices
Class II is divided into class IIa and class IIb. The former includes devices that are middle-risk and the latter – middle- to high-risk.
All devices in this class need to go through the classification process conducted by notified bodies. Only after this process, can they be launched and released onto the market.

Devices from this class can be used in direct contact with a patient and have a therapeutic, diagnostic or monitoring purpose.
When it comes to the market share, class IIa is at around 20% and class IIb – at 8%.
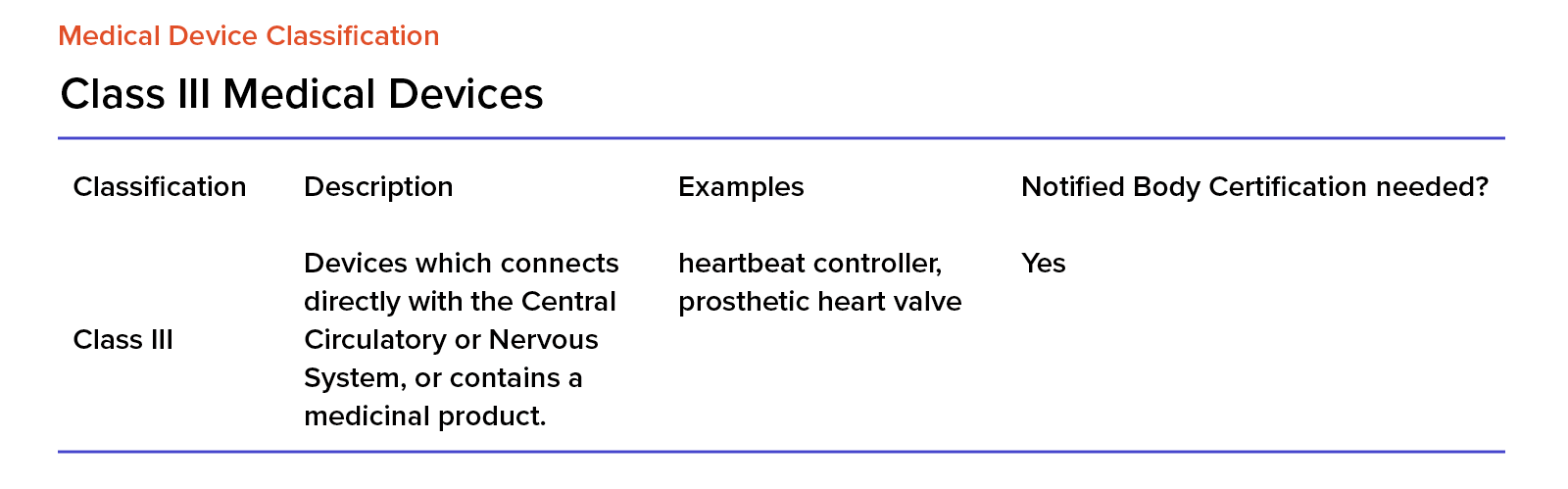
EU MDR Class III Medical Devices
Class III devices are high-risk and when considering implementing and using them, we must ensure that using such a device will have more benefits than risks to the patient. This is particularly important for any device that’s directly connected to a patient’s body.
How is software regulated in the EU MDR?
Software is classified as an active medical device. The MDR now regulates and classifies it, introducing new procedures and requirements that are specific to software. In the MDD, software was in class I, chapter 2 in the MDR has changed it and extended the definition of an active medical device. This makes the whole classification process more of a burden to the manufactures. The software can be a standalone medical device or a part of a system. For class II, it is usually used for measuring, therapeutics or diagnostics.
EU MDR Chapter II – complementing rules
In chapter 2 of the MDR, there are two important rules, crucial to software development. One is that if we have software that’s part of a different device, then it’s in the same class as this device and must be assessed accordingly. If software can be used separately, then it’s classified as a standalone active medical device.
Rule 9
Rule 9 in Annex VIII to the MDR states that if we have a device that’s used for supplying or exchanging energy to/with a human body, then it falls under class IIb.
The same rule applies to devices that can be used for managing and monitoring any therapeutic activities or are directly involved in influencing any devices that can influence the former. Then these devices can also be classified in class IIb.
All active medical devices that can be used to ionise radiation for therapeutic purposes, fall under class IIb. Active medical devices for managing or monitoring, or that affect the radiation directly are in class III.
Rule 10
Rule 10 applies to any active medical device – including software – that can be used for monitoring and diagnostics, automatically land in class IIa. However, if they are used for providing energy that will then be consumed by the human body or for luminating a patient’s body in the visible spectrum, then they fall under class I.
There are certain exceptions to this rule. If medical devices are used for in-vitro imaging the placement of radiopharmaceutical devices or if they are aimed at diagnosing and monitoring essential physiological processes – excluding monitoring devices where there’s a high risk for a patient – in this case, these devices always fall under the class IIb.
Rule 10 also states that when we have an active medical device used for ionising radiation or for radiological diagnostic and therapeutic purposes – including devices that can be used for interventional radiation and devices for managing ad monitoring the former devices or directly affect their activity – then they are in class IIb.
Rule 11
Rule 11 states that software that will be providing information used in the decision-making process for diagnostic or therapeutic purposes falls under class IIa.
The exception to this rule is related to decisions that can cause death or irreversible damage to a patient’s health. In this case, this software is in class III. If decisions could lead to surgical intervention, then class IIb applies.
Software for monitoring physiological processes is in class IIa, unless any changes to these processes can cause irreversible harm to a patient’s health, then they fall under class IIb.
Rule 12
Rule 12 applies to active medical devices that are aimed at supplying and/or removing bodily fluids or other substances to/from a human body, then class IIa applies to them.
If these activities are associated with a higher risk for a patient, then they fall under class IIb.
Rule 15
Rule 15 applies to active medical devices used for contraception or preventing sexually transmitted diseases which usually belong to class IIb. If these devices are used for implantation or are invasive and/or used in the longer term, then they fall under class III.
Rule 22
Rule 22 is associated with active medical devices that can be used for therapeutic purposes, but also have integrated or in-built therapeutic functionalities that heavily influence patient treatment, such as closed flow systems and automatic external defibrillators that fall under class III.
Find out more about the EU MDR from our guide!
Read more
How to assess if your software or your medical product falls under new Medical Device Regulations?
First, we need to define its Intended Use and decide which regulations now apply to your product.
Software is now always an active medical device. If it’s a standalone item, it will be regulated by Annex XIII, Chapter 2 of the MDR.
If it’s a part of a larger device or greatly affects their functioning, then it will be falling into the same class as this product. If this is the case, then please refer to Annex XIII, Chapter 2 of the MDR.
Importantly, some software components can be parts of applications used for certain medical uses. For example, these components can be used for:
- collecting and updating patient data
- storing files containing patient data,
- invoicing and other accounting activities,
- connecting with social security systems,
- connecting with prescription issuing systems,
- connecting with systems that provide expert advice on decision making.
As such, these components do not play any role in medical activities and may be excluded from the classification process. They need – of course – a different approach to system architecture to make this exclusion possible. Spyrosoft can help you with implementing or integrating such a solution.
Summary
To summarise, rule 11 of the EU MDR defines, classifies and formally establishes the requirements for medical devices – including software – and their classification.
Software is no longer classified in class I as it was in the MDD and is now classified in class IIa and IIb.
As a rule, software used for wellness and fitness activities, as well as software for collecting and storing patient data, is not a medical device.
Get expert help with classifying your medical device
Not sure how to classify your medical device? Do not hesitate to contact us using the contact form below – our experts are ready to help.
FAQ
The EU Medical Device Regulation (MDR) came into force in May 2021, replacing the Medical Device Directive (MDD). It was created to improve clinical safety, increase transparency, and unify the regulatory framework for medical devices within the European Union.
Under the MDR, software is now treated as an active medical device, which means stricter requirements for classification, documentation, and clinical evaluation. Software can be standalone or part of a larger system, but in both cases, it must undergo proper risk assessment and compliance verification.
The EU uses four medical device classes: I, IIa, IIb, and III, depending on the level of risk. Software that supports diagnosis or therapy usually falls under Class IIa or IIb, and in some high-risk cases, Class III. The previous MDD often placed software in Class I, which is no longer the case.
Rule 11 (Annex VIII of the MDR) defines how medical software is classified. Software used in diagnostic or therapeutic decision-making is generally Class IIa, but if it can cause serious or irreversible harm, it is classified as Class III. This rule significantly impacts all software manufacturers developing medical applications.
Spyrosoft’s experts can support your company with classification, documentation, and compliance according to the EU MDR. We help define Intended Use, prepare the required technical documentation, and ensure your medical software meets all regulatory standards.




arrow_circle_rightContact us
Need help reclassifying your medical product?

arrow_circle_right our ARTICLES




