What is the EU MDR?


The MDR (Medical Device Regulation) is a new regulation governing the production and distribution of Medical devices. It entered into force on May 26 2021 and applies to all Medical device manufacturers who want to introduce their products on the EU market.
Is there a transition period for adopting the EU MDR?
The EU MDR specifies the transition period for class I devices as four years (until May 26, 2025).
However, it’s important to note that the transition period applies only to manufacturers of devices that are present on the market and only until any changes to the product are implemented.
>> To learn what is considered a change, read this blog post
What are the most important changes introduced by the EU MDR?
The new regulation puts a lot of emphasis on unifying the market. No existing requirements have been removed. However, many have been tightened up, and a few new ones have been introduced.
The key changes include:
Stricter Medical device classification rules
The new, more rigid rules affect especially the manufacturers of invasive devices intended for implantation, surgeries and other devices described as active, including software used with such devices. The most serious change affected Medical devices that were in class 1 under the MDD. Now, they are at least in class 2a.
Changes to the quality management system
The scope of the Quality Management System has been expanded and now also includes, for example, the procedure for clinical evaluation and running a Post-Market Surveillance (PMS) system.
Supervision of notified bodies
There are more requirements concerning the designation of notified bodies, which are now controlled by national competent authorities and the European Commission.
Introduction of an independent expert panel
The MDR introduces a consultation procedure conducted by an independent expert panel for certain class IIb devices (not obligatory) and class III devices intended for implantation (obligatory).
For all devices in classes IIa, IIb and III, the notified body now needs to be involved in the conformity assessment of a product for CE marking.
More rigid clinical evaluation requirements
The new requirements include the collection of clinical data that is already available in the literature and the organisation of the necessary clinical trials.
With certain exceptions, implantable Medical devices and class III Medical devices must now undergo clinical trials. For all class III and IIb devices intended to manage a drug (in or out of the body), the manufacturer may consult a group of EU experts to get an opinion on the clinical development plan.
Better traceability of Medical devices
A new system of Unique Device Identification (UDI) was introduced to improve the identification and traceability of Medical devices.
Increased transparency
The information about products and tests is now accessible to the public through a new database of Medical devices available in the European Union, called EUDAMED.
>> For a more detailed overview of the changes introduced by the EU MDR, read this post
Our experts will help you ensure compliance with EU MDR
Find out more
How to determine if your software qualifies as a Medical device under the EU MDR?
According to the EU MDR, software in its own right, when intended to be used for one or more Medical purposes, qualifies as a Medical device. However, software for general purposes, even when used in a healthcare setting or for lifestyle and well-being purposes, is not considered as a Medical device. The key factor in determining whether the software is considered a Medical device or not is its Intended Use.
As a side note, let’s add that the Intended Use is a document that specifies the predetermined usage of a certain Medical device. It is necessary to prepare a certification strategy and assess the class of a product.
Based on your software’s Intended Use, you can now determine whether or not your software is a Medical device following these guidelines:
Software is a Medical device if:
- is explicitly named as one in the EU MDR
- controls or influences a Medical device
- serves the “post-processing” (e.g., for an ECG) or data preparation
- calculates output signals or output values
- serves the support of diagnostic or therapy
- serves the “objective” diagnosis or treatment
Software is not a Medical device if:
- is used for administrative purposes (e.g., managing patient data)
- is used for training doctors (e.g., training software with Medical knowledge)
- is used for general maintenance of Medical devices or their components (however, sometimes can be treated as a Medical device accessory)
- is used for development or production tools (however, such must be validated)
- represents a proprietary operating system
>> Here you can see some examples of qualifying Software as a Medical Device
How to classify a Medical device under the EU MDR?
If you determined that your software is considered as a Medical device according to the EU MDR, the second step is to determine which class it qualifies for.
Firstly, it’s important to note that the classification of a device should be separated from the classification of software that may be a part of this device. In other words, software, regardless if it’s a part of a device or a standalone product, needs to be classified separately.
Is it possible that a Medical device and software have different classes? Yes, it’s possible. For example, a class III Medical device that has the highest risk level can be supported by software that includes only simple, basic functionalities, doesn’t impose any risk, and therefore qualifies to a lower class than the Medical device. Additionally, according to IEC 62304, it’s possible to declassify certain software components if specific criteria considering isolation and decomposition are met.
MDR has 22 rules that determine how the classification should be conducted. There are currently four Medical device classes used in the European Union: I, IIa, IIb and III, which depend on the risk level associated with a device.
Let’s move on to the classification process itself.
The first step is to ensure that we have the necessary documents and information about the Medical device to be classified. The basic ones that need to be prepared and collected before the classification process are:
- device description, including its Intended Use, preferred usage and user group,
- data on the user group described as patients and on the diseases that can be affected by the device, the treatment, diagnostics and monitoring.
Based on this information, you can assess your device’s class.
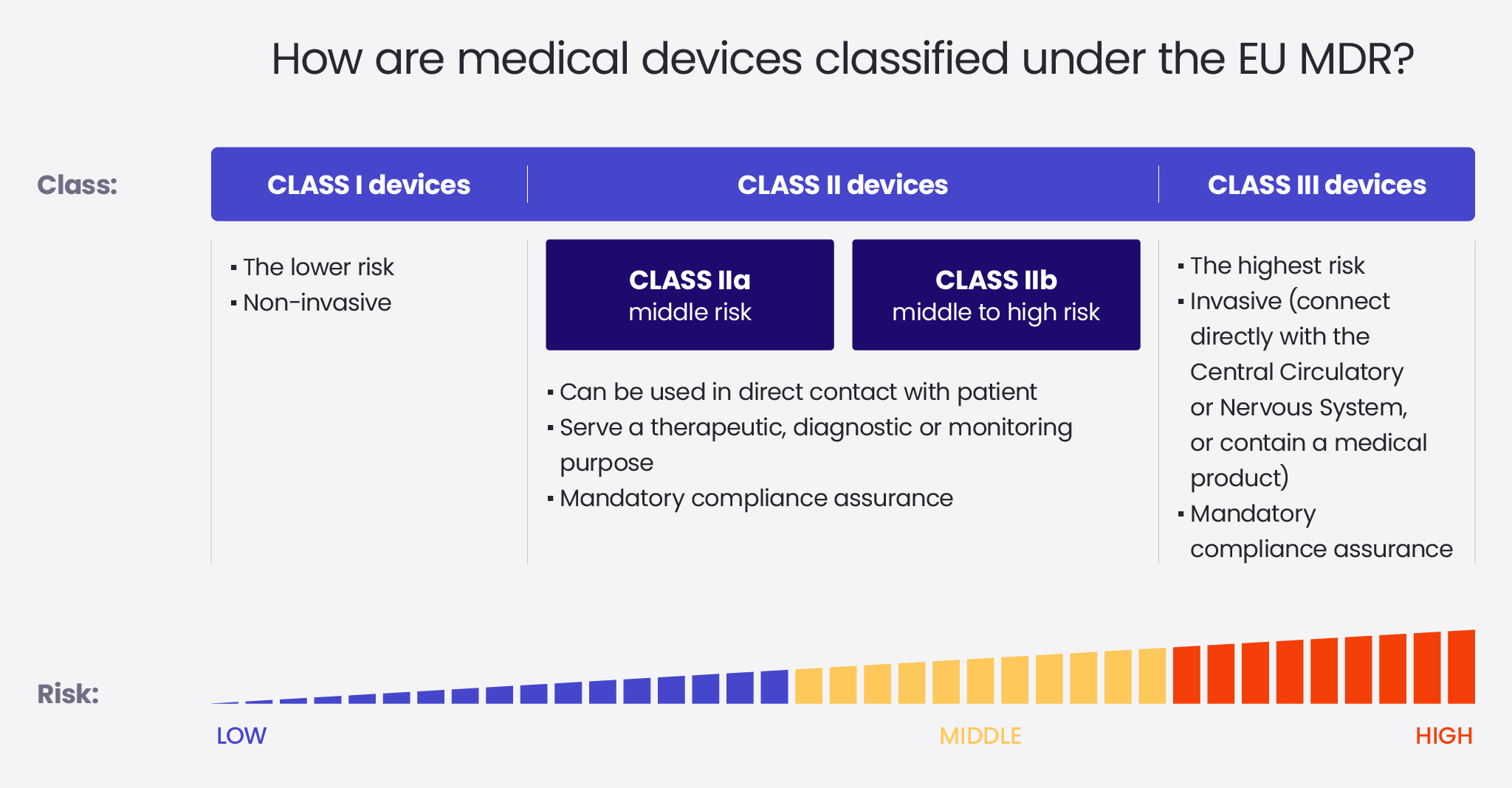
Class I
Class I is associated with the least risk. Devices that belong to this class cannot be invasive, and they cannot be in direct contact with a patient.
Class II
Class II is divided into class IIa and class IIb. Class IIa includes devices that are middle-risk and class IIb – middle- to high-risk. Class II devices can be used in direct contact with a patient and serve a therapeutic, diagnostic or monitoring purpose. Class II Medical devices have to go through a compliance assurance process conducted by a notified body.
It’s crucial to keep in mind, that according to the new regulation, any software is now classified by default to, at least class IIa. It has important implications for the software development process, which has become more complex and thus requires more time.
Class III
Class III devices are associated with the highest risk. They are described as those which connect directly with the Central Circulatory or Nervous System, or contain a medicinal product, for example, a heartbeat controller or a prosthetic heart valve. Class III Medical devices, similarly to class II Medical devices, have to go through a compliance assurance process conducted by a notified body.
>> You can find more detailed information about each class in this article
Do you have questions about the implementation of the EU MDR?
If you’re still unclear about the EU MDR requirements or the classification process for your software, check this list of the most puzzling questions concerning the EU MDR that we came across.
You can also contact us for assistance in implementing the EU MDR requirements to your software. We’ll guide you through the process and help with conducting an independent audit to identify areas that need improvement as well as propose optimal solutions.
See our full offer or contact us through the form below.




arrow_circle_rightContact us
Need help reclassifying your medical product?

arrow_circle_right Our articles



