A guide to FDA medical device regulations


From this guide, you’ll find out:
- what the process for releasing a medical device in the U.S. looks like step by step,
- what requirements for medical device manufacturers are set out in the regulations,
- how to determine if your product qualifies as a medical device and how to classify it properly,
- what submission method should you follow and why,
- how to comply with both MDR and FDA to sell your device on both the EU and US markets.
How to bring your medical device to the U.S. market?
The process of bringing a medical device to the U.S. market, as determined by the FDA, can be broken down into the following main steps:
- Determine if your device is considered a medical device based on its Intended Use and the Indications for Use.
- Classify the device. Class I medical devices, while subject to less stringent requirements, must still comply with basic guidelines including device registration.
- Prepare scientific evidence that your medical device serves its role as defined in the Intended Use.
- Choose the appropriate premarket submission pathway.
Let’s dig deeper into each step of the process.
FDA Medical Device Definition
The first step in the process is to validate your product against the FDA definition of a medical device, which is included in Section 201(h) of the FD&C Act (21 USC 321(h)).
The FDA defines a medical device as an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article that is intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease, or intended to affect the structure or any function of the body. Understanding this definition is crucial for manufacturers to determine if their product qualifies as a medical device and to ensure compliance with FDA regulations.
Our experts will help you achieve compliance with FDA’s regulations
Learn more
How to determine if your product is a medical device, and how to classify it according to the FDA’s regulations?
Navigating through FDA classification regulations helps identify the appropriate category among the 16 medical specialties, ensuring compliance with regulatory requirements.
The easiest method to validate if your product meets the criteria specified in the medical device definition above is to identify its Intended Use(s) and the indications of using it. Once you have them documented, you can move on to the next step of the validation process: checking for appropriate product classification. Understanding whether a device falls under the FDA’s specific definitions and classifications is crucial for proper categorisation. There are at least three methods to do that:
1. Product Classification Database search
Visit the FDA Product Classification Database to determine whether there’s an existing classification your product may fit into. Please note that you may need to use both Quick Search and Advanced Search and enter at least a few search keywords until finding the right ones.
2. Comparing similar devices
If you know any devices that are similar to your product, you can type, for example, its name, code, device class and regulation number, and base your product’s identification on the results. If you don’t know any products that may fit into the same category, you can also look through databases that are created for premarket devices in low to moderate risk class, such as Premarket Approval (PMA), Premarket Notification 510(k), and De Novo. There’s also the Humanitarian Device Exemption (HDE) for devices used for diagnosing, curing and mitigating rare diseases or conditions.
3. Device listing information
Visit the Establishment Registration and Device Listing database and check for medical device manufacturers registered with the FDA and the products they release onto the market.
>> How to determine if your product is a medical device according to the FDA?
What are the medical device classes?
The classification process is based on risk assessment and regulatory controls necessary to ensure patient and operator safety, as well as the effectiveness of a device. According to the FDA, there are three medical device classes:
- Class I – associated with the lowest risk level.
- Class II – posing a moderate risk to users. Examples of Class II devices include blood transfusion kits, which require stringent FDA regulation to ensure their safety and effectiveness.
- Class III – characterising devices with the highest risk level. Class III devices, such as implantable pacemakers, require FDA premarket approval (PMA) due to their high-risk nature and the need for extensive clinical trials.

>> How to classify software as a medical device according to the FDA and FDA Level of Concern: An Introduction to the FDA’s Regulation of Medical Devices
Overview of 21 CFR
Title 21 of the Code of Federal Regulations (21 CFR) is a comprehensive set of federal regulations that govern the development, testing, and marketing of medical devices in the United States. These regulations are divided into several parts, each addressing a specific aspect of medical device development and regulation. 21 CFR provides a framework for ensuring the safety and effectiveness of medical devices, and compliance with these regulations is mandatory for all medical device manufacturers. By adhering to these federal regulations, manufacturers can ensure that their products meet the necessary standards for safety and efficacy before they are marketed.
Electronic Records and Signatures (21 CFR Part 11)
21 CFR Part 11 regulates the use of electronic records and signatures in the development and manufacture of medical devices. This part of the regulation requires that electronic records be accurate, reliable, and secure, and that electronic signatures be authentic and verifiable. The regulation also mandates that electronic records be maintained in a way that ensures their integrity and authenticity, and that they be readily available for inspection by FDA officials. Compliance with Part 11 is essential for manufacturers who use electronic systems in their processes, as it ensures that all records are trustworthy and can be relied upon during regulatory reviews.
What is a 513(g) Request?
If you have difficulties with the classification of your medical device, you can ask the FDA for a formal device determination or classification by submitting a 513(g) Request. However, this option is additionally paid – a standard fee is $5,061 – and the process takes a substantial amount of time.
If you need help with the classification of your device, our expert can help you at a similar cost and in a much shorter time. Contact us using the form below for more information.
How to check if your product is Software as a medical Device (SaMD)?
If your product is a stand-alone application that may or may not be used as a part of a larger device, it may be classified as Software as medical Device (SaMD).
Follow this link to learn more about SaMD and establish if your product can be classified as such: Software as a medical Device: what you need to know in 2022
Types of the FDA’s premarket submissions
The class of a device determines the type of submission for clearance to market required by the FDA. There are five types of submission:
Premarket Notification 510(k)
510(k) is used for class I devices and the majority of class II devices, which aren’t subject to another kind of premarket submission.
Premarket Approval (PMA)
It’s the most stringent type of premarket submission required by the FDA. It is applied to class III devices that present the highest risk or don’t have substantial equivalents already available on the market.
Investigational Device Exemption (IDE)
IDE applies to investigational devices that are to be used in clinical studies to collect data on their safety and effectiveness and to identify potential risks to human health and life.
De Novo classification request
De Novo classification request is a submission pathway for novel class I or class II devices with no similar predecessors available on the U.S. market which don’t fit any existing categories.
Humanitarian Device Exemption (HDE)
HDE is a premarket submission method for class III devices presenting a high-risk level. HDE usually applies to devices intended to treat rare diseases or medical conditions.
>> Read about the types of the FDA’s premarket submissions in more detail in this article: An introduction to the FDA’s regulation of medical devices
Pre-Market Approval for Class III Medical Devices
Class III medical devices are the most stringently regulated devices, and they require pre-market approval (PMA) from the FDA before they can be marketed. The PMA process involves the submission of a detailed application to the FDA, which includes comprehensive information about the device’s design, testing, and clinical trials. The FDA reviews the application to ensure that the device is safe and effective, and that it meets all regulatory requirements. The PMA process can be lengthy and complex, often requiring significant resources and expertise. Manufacturers of Class III medical devices must be prepared for a rigorous review process to demonstrate that their products are safe for use.
510(k) Clearance for Class II Devices
Class II medical devices require 510(k) clearance from the FDA before they can be marketed. The 510(k) process involves the submission of a pre-market notification to the FDA, which includes detailed information about the device’s design, testing, and intended use. The FDA reviews the notification to ensure that the device is substantially equivalent to a predicate device that is already legally marketed. This process is generally less complex and time-consuming than the PMA process for Class III devices, but it still requires significant resources and expertise. By obtaining 510(k) clearance, manufacturers can demonstrate that their Class II devices meet the necessary regulatory standards for safety and effectiveness.
The FDA’s requirements regarding the quality management system
The requirements regulating how a quality management system should be designed and developed to ensure a medical device is safe to use are described in Part 820 of the FDA’s CFR Title 21.
It’s worth noting that the requirements of Part 820 and ISO 13485 are almost equivalent, so if your quality management system is already compliant with ISO 13485, it’s likely to also be in line with Part 820.
How much does it cost to get FDA approval for a medical device?
For most of its applications for medical device review, the FDA charges a fee that must be paid to start the approval process unless the applicant is eligible for exemption. Besides the application costs, the FDA also charges an Annual Establishment Registration Fee, which is $5,672 for 2022.
It is worth mentioning that there is a chance of qualifying for reduced fees for small businesses. The fees for the types of applications listed in the text are as follows:
How long does FDA premarket approval take for medical devices?
The time it takes to get a medical device approved depends on its class and can last from one or two weeks to eight months.
>> Read more about FDA approval process for medical devices
EU MDR vs. FDA: what are the main differences and similarities?
There are several differences between the FDA and MDR requirements, but also some similarities in certain areas. Important ones are listed below.
Medical device classification
The FDA categorises medical devices marketed in the United States into three classes based on their risk profile, affecting the marketing regulations and requirements for manufacturers.
The approach to medical device classification under the FDA is quite similar to the MDR. Under the EU MDR, there are classes I, II (further divided into IIa and IIb) and III. The higher the class – the higher the risk.
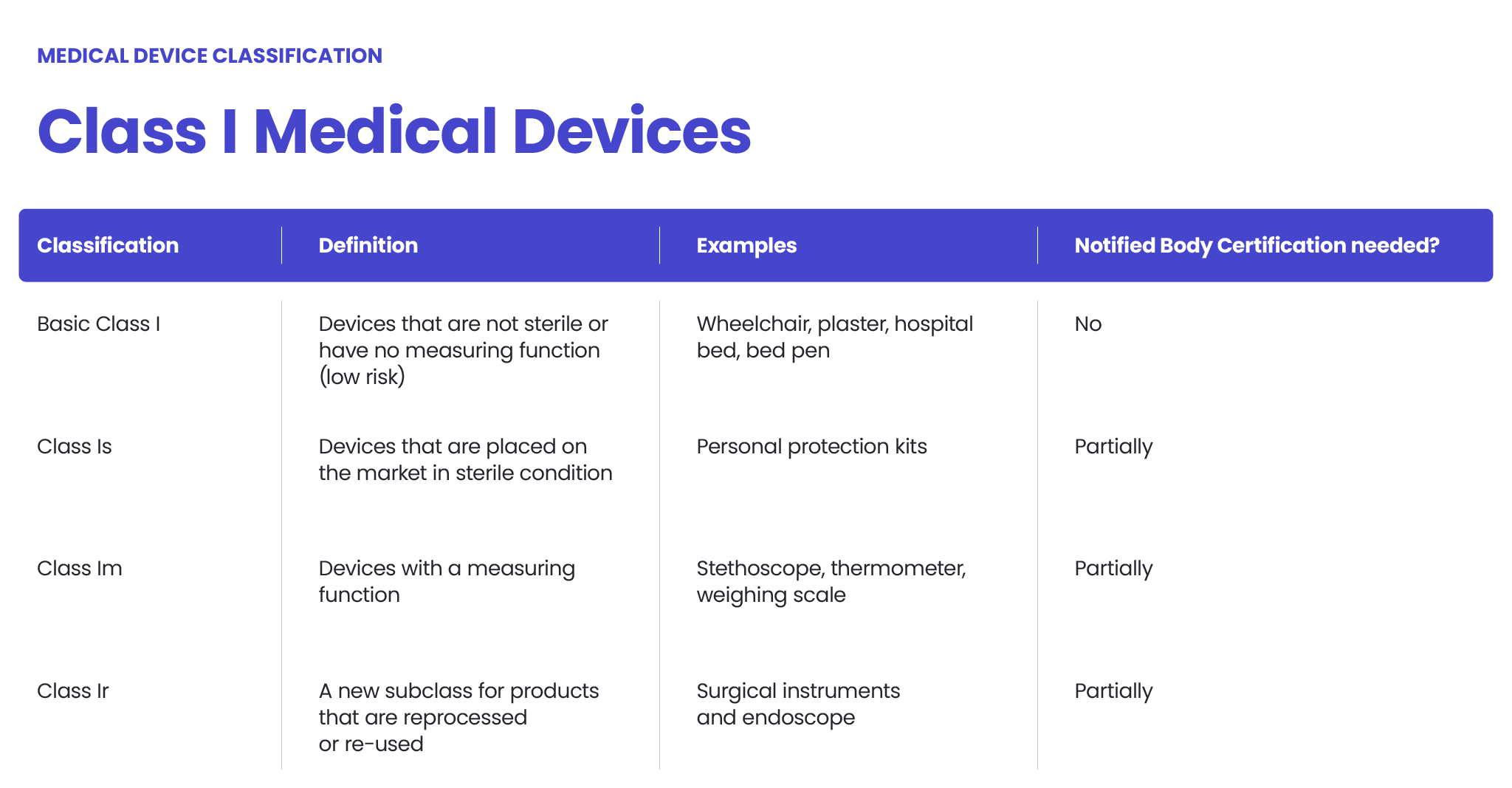

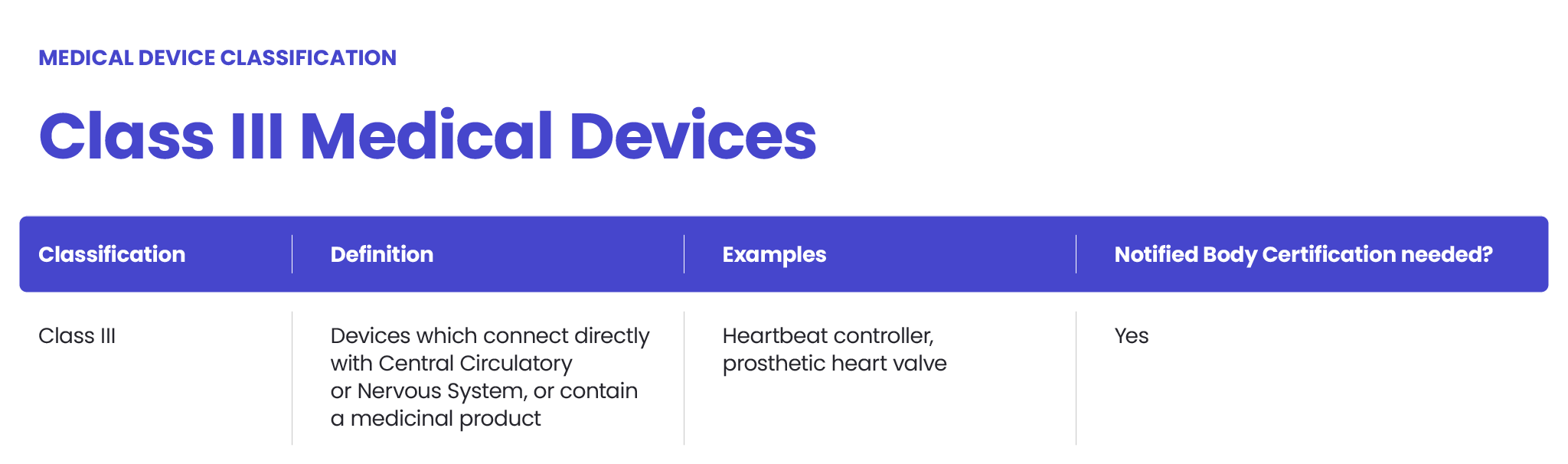
The FDA also classifies medical devices according to their risk level and regulatory controls necessary to ensure patient and operator safety: class I is associated with the least risk, class II with moderate risk and class III with the highest risk.
However, there may be differences between the MDR and FDA in assessing the risk level. The MDR Class I sterile or measuring devices may be regarded as class II under the FDA’s regulation. Certain MDR class IIb medical devices could be regarded as class III, according to the FDA. Each case should be assessed independently.
Quality management systems
Both the MDR and FDA require manufacturers of medical devices, including software, to have a quality management system implemented that is compliant with respective regulations.
The scope of content
The FDA regulations are divided into sections according to the categories of medical devices. Each section includes requirements regarding the safety and effectiveness of medical devices as per their use. It’s directly connected to the premarket submission method followed later in the process.
The MDR focuses more on the specific requirements for medical device manufacturers and notified bodies who overview the process of introducing new medical devices to the EU market.
Submission process
According to the FDA and MDR, class I medical devices (of the lowest risk, non-measuring and non-sterile ones) are not required to undergo an independent audit before their release to sale. To introduce them to the EU and US markets, the manufacturers must follow a self-declaration process (in the EU, it’s a CE-marking process). Naturally, all the appropriate documentation must be prepared, but third-party verification is not required.
Medical devices of higher risk must be verified either by a notified body in the EU or by the FDA in the US. A Technical File must be prepared in both cases, even though the release-to-market process is different.
>> EU MDR vs FDA: what are the main differences and similarities?
Medical device databases
There are different medical device databases functioning in the EU and the US. In the US, it’s the FDA’s medical device database and in the EU – EUDAMED.
Unique medical device identification system
UDI is a globally accepted system and applies in the EU as well as in the US. However, there are certain differences between the MDR and FDA as to where and how the labels should be placed on medical devices.
Technical documentation
Despite the fact that the FDA and MDR regulations regarding the Technical File bear certain similarities, it doesn’t mean that the Technical File created for the EU requirements will be equally acceptable in the US and vice versa. Some differences lie in the nuances regarding manufacturing medical devices, such as risk management and classification.
In order to comply with the FDA and MDR requirements, your documentation should cover the requirements specified in both regulations. It’s not only the best possible but also recommended approach.
Clinical evaluation
For all class III and some class IIb medical devices, the EU MDR requires a Clinical Evaluation Report (CER) to be prepared by a manufacturer and then reviewed and accepted by a Notified Body. In contrast, the FDA doesn’t require CER for most medical devices qualified for a 510(k) submission.
The usability of medical devices
Both the FDA and the EU MDR put a strong focus on the usability of medical devices.
We’ll support you in releasing your medical software onto the U.S. market
Our experts in the FDA requirements for medical devices will guide and assist you throughout the whole process. We will also assist you in preparing all the necessary technical documentation to meet the requirements of both the EU and US medical device markets. Check our FDA regulatory consulting services.
See also our Healthcare and Life Sciences offering page for more details about our services, or contact us directly using the form below.
FAQ: FDA medical device regulations
Determining whether your product meets the U.S. legal definition of a medical device under Section 201(h) of the FD&C Act. This depends mainly on your product’s Intended Use and Indications for Use. If your product is intended to diagnose, treat, cure, mitigate, or prevent disease (or affect the structure or function of the body) it may qualify as a medical device.
It’s is a formal submission to the FDA asking for classification guidance. It involves a fee and can take time to process, but it provides official confirmation of how your device is regulated.
A 510(k) submission demonstrates that your device is substantially equivalent to a legally marketed device (predicate device). It is the most common pathway for Class II devices.
If your product is standalone software intended for medical purposes (diagnosis, treatment, monitoring, etc.), it may qualify as Software as a Medical Device (SaMD). These products must follow the same classification and regulatory pathways as other medical devices.
Title 21 CFR (Code of Federal Regulations) governs the development, testing, manufacturing, and marketing of medical devices in the U.S.
It includes requirements for:
– Quality systems
– Labelling
– Electronic records (Part 11)
– Premarket submissions
Compliance is mandatory for manufacturers.




arrow_circle_rightContact us
Seeking assistance with the FDA approval of medical devices?

arrow_circle_right Our articles



